1. Die Entdeckung des Gen-Ortes
2. Vererbung
3. Welche Auswirkung & Funktion hat das verantwortliche Gen MeCP2
1. Die Entdeckung des Gen-Ortes
1966 publizierte Prof. Rett seine Daten zu 21 Mädchen mit sehr ähnlichen Kernsymptomen, der seither als Rett Syndrom bekannten Erkrankung. Erst 33 Jahre später, 1999, wurde das dafür verantwortliche Gen, das MECP2 Gen, gefunden.
Warum ist dies so schwierig: Die 23 menschlichen Chromosomenpaare tragen mehr als 100.000 Gene. Jedes Gen besteht wiederum aus unterschiedlich vielen Nukleinsäuren. In Summe ist das wie die Suche nach der Nadel im Heuhaufen oder auch die Suche nach einem Tippfehler in einem sehr dicken Buch. Nun wusste man aus den klinischen Beschreibungen, dass primär nur Mädchen betroffen sind, damit also die Suche auf das X-Chromosom oder, um bei dem Vergleich mit dem Buch zu blieben, auf das Kapitel „X-Chromosom“ beschränkt werden kann. Erschwert wurde die Suche dadurch, dass man nur sehr vereinzelt familiäre Fälle mit mehreren betroffenen Mädchen kannte, da die allermeisten Fälle (mehr als 99.9%) spontan auftreten und keine weiteren betroffenen Familienmitglieder vorliegen.
Interessanterweise kannte man das Gen MECP2 schon seit Jahren, aber nicht den Zusammenhang mit dem Rett Syndrom. Hier gab es auch eine geografische Nähe. Prof. Rett war ja bekannter weise am Neurologischen Zentrum Rosenhügel aktiv, Prof. Adrian Bird arbeitete Anfang der 90er Jahre ebenfalls in Wien an einem molekularbiologischen Zentrum an genau diesem MECP2, jedoch ohne von diesem späteren Konnex zu wissen.
Schließlich halfen Daten aus großen Datenbanken und entsprechende Familien mit mehreren Betroffenen, den Standort der betroffenen Region von einigen tausend auf einige hundert Gene zu reduzieren. Die beiden Gruppen um Huda Y. Zoghbi und Uta Francke engten dann die möglichen Gene Schritt für Schritt ein, bis 1999 die Publikation den kausalen Zusammenhang beschreiben konnte.
2. Vererbung
Wie bereits eingangs erwähnt, tritt die Mutation (genetische Veränderung) bei den betroffenen Mädchen „neu“ auf (auch „de-novo“ genannt). Dies bedeutet, dass die genetische Veränderung/Mutation meist einmalig und zufällig am X-Chromosom der Keimzelle (Spermien) stattfindet und damit an die Nachkommen weitergegeben wird. Die Töchter haben generell 2 X-Chromosomen, Söhne ein X- und ein Y-Chromosom. Deshalb erkranken Söhne ausnahmslos (sehr schwer und früh), Töchter zeigen das Bild eines klassischen Rett Syndroms. Dass es sehr seltene familiäre Fälle von Geschwistern mit Rett Syndrom und unterschiedlichem Schweregrad gibt, hat mit der Tatsche zu tun, dass normalerweise und in den allermeisten Fällen eines der beiden X-Chromosomen beim Mädchen funktionell ganz früh in der Embryonalentwicklung stillgelegt wird. In diesen seltenen Fällen kommt es zu einer „schrägen“ Inaktivierung, also nicht 50:50, und damit mehr oder weniger betroffenen Körperzellen und unterschiedlich schwere klinische Symptome.
Das hier Beschriebene gilt nicht für Burschen mit Rett Syndrom und einer Duplikation von MeCP2, dies ist ein genetisch und klinisch differentes Bild.
Bleiben wir beim „Normalfall“: Es wurde eine MeCP2-Muation bei einem Mädchen gefunden:
Wenn diese von der Patientin bekannte Mutation weder bei der Mutter noch beim Vater gefunden wird, dann ist von einer de-novo Mutation auszugehen. Außer es besteht ein Gonaden-Mosaik – siehe oben –, das jedoch mittels Bluttest nicht untersuchbar ist. Dann besteht kein erhöhtes Wiederholungsrisiko für weitere Kinder im Vergleich zur Wahrscheinlichkeit gegenüber der Normalbevölkerung ohne Kind mit Rett Syndrom. Bei einer weiteren Schwangerschaft kann jedoch nach Beratung eine gezielte, pränatale molekulargenetische Diagnostik dieser bei der betroffenen Schwester gefunden Mutation durchgeführt werden.
Gesunde Geschwister eines Mädchens mit RTT tragen keine MECP2-Mutation. Aus diesem Grund ist auch das Risiko bei eigenen Kindern nicht erhöht. Ausnahmen bestehen nur bei den sehr seltenen Fällen, die auf Basis einer „schrägen“ X-Inaktivierung weitervererbt werden.
Prinzipiell kann in den Familie von Mädchen mit Rett Syndrom und bekannter MECP2 -Mutation (bei 5-10% der Mädchen findet man keine kausale Veränderung im Gen) diese bei Familienmitgliedern auch untersucht werden. Diese Untersuchung/en sollten jedoch nur nach vorangegangener, eingehender genetischer Beratung erfolgen.
3. Welche Auswirkung & Funktion hat das verantwortliche Gen MECP2
Jedes unserer Gene (also „Buch-Kapitel“ des gesamten Genoms) repräsentiert den Bauplan eines ebenso bestimmten Eiweiß-Stoffes (Proteins). Im Falle des MeCP2 Gens ist dieses für das MeCP2 Protein verantwortlich. Dieses Eiweiß wird in vielen Körpergeweben produziert und benötigt.
Prinzipiell hat jedes Gen, so auch das MECP2 Gen einen genauen „Fahrplan“, wann es im Laufe der Embryonalentwicklung und des Lebens aktiv ist und wann es „abgeschaltet“ ist. Daraus ergibt sich neben dem genetischen Bauplan des Menschen (Gene) auch ein genetischer Zeitplan.
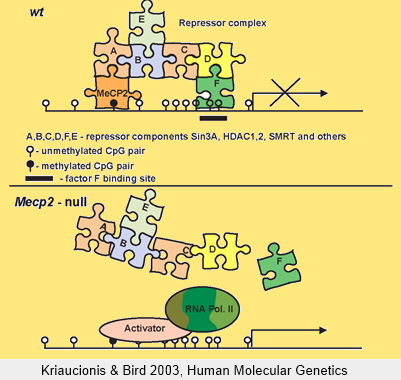
Das MeCP2 Protein hat nicht eine einzelne Aufgabe wie zum Beispiel Eiweiß-Enzyme, sondern dürfte für die übergeordnete Regulation anderer Gene verantwortlich sein, derer bereits einzelne gut untersucht sind.
Wie diese Abbildung veranschaulichen soll, ist das MeCP2-Protein ein wichtiger Bestandteil oder Puzzle-Stein eines Komplexes, der ohne MeCP2 (siehe unterer Teil der Abbildung) nicht funktioniert und zu einer veränderten Regulator-Funktion führt.
MECP2 wird in sehr vielen Forschungslabors rund um den Globus beforscht, weil es aufgrund des Gesagten ein sehr interessantes Gen ist. Interessant vor allem wegen seiner Bedeutung für die Gehirnentwicklung und die Regulator-Funktion.
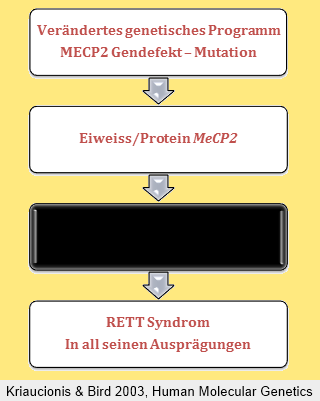
Wie die Grafik veranschaulichen soll, wissen wir sehr viel über das Gen MECP2 und das Protein MeCP2, aber wir wissen noch (zu) wenig über die genauen Funktionen, die dann zu den bekannten Symptomen führen. Viele therapeutische Ansätze setzen hier am MeCP2 Protein an (siehe Bericht von F. Laccone im letzten Rundbrief oder auch unsere Creatin-Studie). Neue Möglichkeiten der Beeinflussbarkeit werden sich möglicherweise durch neue Erkenntnisse über diese black box ergeben.
OA Dr. Michael Freilinger
Neuropädiatrie Universitätsklinik f. Kinder- u. Jugendheilkunde
Medizinische Universität Wien – AKH Wien
Email michael.freilinger@meduniwien.ac.at
zurück zur FORSCHUNG